Редкий синдром Марфана: признаки, способы лечения и наследование у детей. IV. Синдромы Марфана, Морриса и андрогены Известные люди с синдромом марфана
Врожденные заболевания костной и мышечной систем органов и тканей человека
С. Ю. АФОНЬКИН
Дефекты некоторых генов, влияющих на образование и развитие соединительной ткани у человека, нередко приводят к непропорциональному гигантизму. Болезнь эта была описана в 1896 г. французским педиатром А. Марфаном.
Антуан Марфан, французский педиатр
При наиболее ярком проявлении этой доминантной особенности на свет появляются люди с очень длинными руками и ногами и относительно коротким туловищем. Их вытянутые пальцы напоминают лапы огромного паука. Отсюда образное название этой диспропорции - арахнодактилия (от греч. dactyl - палец и Arachna - женщина, по легенде, превращенная Афиной в паука). Люди с подобными дефектами необычайно худы, их грудная клетка бывает деформирована, хрусталик глаза смещен.
Такая аномалия носит название синдрома Марфана и считается полулетальной, поскольку связана с пороками сердца. Синдром вызван наследственным пороком развития соединительной ткани и характеризуется также поражением опорно-двигательного аппарата, глаз и внутренних органов. Первопричины таких пороков недостаточно изучены.
Нередко люди с арахнодактилией умирают от аневризмы аорты - самый крупный сосуд, выходящий из правого желудочка сердца, не выдерживает давления выбрасываемой в него крови. Люди, у которых этот синдром проявляется не со всей жестокостью, доживают до зрелых лет. По счастью синдром Марфана встречается достаточно редко. Специалисты оценивают вероятность его появления как 1/50000.

Единственная компенсация, которую люди с синдромом Марфана получают от судьбы за свой порок, - повышенное содержание адреналина в крови. Как известно, этот гормон вырабатывается надпочечниками и выбрасывается в кровяное русло в момент опасности. В результате многие параметры человеческого организма (сердцебиение, давление крови) приводятся, так сказать, в боевую готовность. Таким образом, люди с синдромом Марфана всю жизнь находятся в возбужденном состоянии: адреналин постоянно подстегивает нервную систему и делает их невероятными трудоголиками.
Синдромом Марфана страдали несколько всемирно известных личностей, отличавшихся необычайной работоспособностью. Таков был лесоруб Авраам Линкольн, который благодаря постоянному самообразованию, выдающимся способностям и, главное, потрясающему трудолюбию стал президентом США. Он обладал высоким ростом - 193 см, огромными стопами и кистями рук, маленькой грудной клеткой и длинными гибкими пальцами - типичное телосложение при синдроме Марфана.

Авраам Линкольн
Очень похож на Линкольна по физическому складу был сын полунищего сапожника, ставший позже великим писателем XIX в., Ханс Кристиан Андерсен. Его необычайное трудолюбие проявилась еще в школе. Свои литературные произведения он переписывал до десяти раз, добиваясь в конечном счете виртуозной точности и одновременно легкости стиля.

Ханс Кристиан Андерсен
Современники так описывали его внешность: «Он был высок, худощав и крайне своеобразен по осанке и движениям. Руки и ноги его были несоразмерно длинны и тонки, кисти рук широки и плоски, а ступни ног таких огромных размеров, что ему, вероятно, никогда не приходилось беспокоиться, что кто-нибудь подменит его калоши. Нос его был так называемой римской формы, но тоже несоразмерно велик и как-то особенно выдавался вперед».
Нервное напряжение, в котором, по-видимому, постоянно находился этот талантливый человек, порождало у него множество страхов. Он боялся заболеть холерой, пострадать от пожара, попасть в аварию, потерять важные документы, принять не ту дозу лекарства...
История знает случай, когда длинные, тонкие пальцы человека с синдромом Марфана вместе с впечатляющей работоспособностью помогли их обладателю сделать фантастическую карьеру. Речь идет о знаменитом скрипаче Никколо Паганини. Гете и Бальзак так описывают его внешность в своих воспоминаниях: мертвенно-бледное, как будто вылепленное из воска лицо, глубоко запавшие глаза, худоба, угловатые движения и, самое главное, тонкие сверхгибкие пальцы, какой-то невероятной длины, как будто вдвое длиннее, чем у обычных людей. Эта чисто морфологическая особенность позволяла ему творить со скрипкой настоящие чудеса.

Никколо Паганини
В толпе, слушавшей импровизации Паганини на римских улицах, одни говорили, что он в сговоре с дьяволом, другие - что его искусство является музыкой небес, в которой звучат ангельские голоса. Он играл так, что слушателям казалось, будто где-то спрятана вторая скрипка, играющая одновременно с первой. Многие вплоть до XX в. верили слухам, что в молодости Никколо прибег к помощи хирурга, который сделал ему операцию, чтобы повысить гибкость рук. Теперь-то мы знаем, что, скорее всего, своим данными он был обязан редкому генетическому отклонению.
Впервые на связь мастерства Паганини с синдромом Марфана указал американский врач Майрон Шенфельд в статье, опубликованной в «Журнале Американской медицинской ассоциации». Он указал, что описание внешности Паганини - бледная кожа, глубоко посаженные глаза, худое тело, неловкие движения, «паучьи» пальцы - абсолютно точно совпадает с описанием облика людей с синдромом Марфана. Как известно, в конце своей жизни великий музыкант почти лишился голоса. Это лишнее свидетельство в пользу того, что у Паганини был синдром Марфана, поскольку нередким осложнением этой болезни является сильная хрипота или даже потеря голоса, вызванная параличом верхнего гортанного нерва.
Сохранился дневник врача, который лечил Паганини. Сделанные в нем записи подтверждают классические симптомы синдрома Марфана: астеническое сложение, явно выраженный сколиоз, «птичье» выражение лица, узкий череп, выступающий или срезанный подбородок, глаза с синими склерами, разболтанность суставов, диспропорции в величине туловища и конечностей, кисти и стопы длинные с тонкими «паукообразными» пальцами. Не удивительно, что не только игра Паганини, но и сама его необычная внешность производили впечатление на его современников, рождая подчас самые немыслимые легенды о музыканте.
Надо заметить, что сам по себе синдром Марфана не располагает к музыкальной одаренности. За исключением Паганини, среди больных с этим синдромом не было выдающихся музыкантов. Что же касается Паганини, то болезнь лишь придала ему большие технические возможности, а великим музыкантом, с огромным творческим наследием, включающим, кроме произведений для скрипки с другими инструментами и оркестром также более 200 пьес для гитары, он стал благодаря своему великому таланту и трудолюбию, тоже косвенно связанному с синдромом Марфана.
Наверняка вы припомните еще двоих знаменитых длинных, нескладных и талантливых «носачей». Это Шарль де Голль и Корней Иванович Чуковский. Деятельный характер будущего президента Франции настолько ярко проявлялся еще в молодости, что многие из его сослуживцев по армии до Второй мировой войны уже тогда прочили его в генералиссимусы. Голова де Голля всегда возвышалась над морем касок и беретов марширующих солдат. Вместе с тем, сидя за столом, он казался вполне обычным человеком. Секрет крылся в его непропорциональном сложении, столь характерном для синдрома Марфана.

Шарль де Голль
Выше всех в толпе был и любимый детьми автор «Мухи-цокотухи», «Мойдодыра» и «Тараканища». Его длиннорукость, длинноногость, большеносость и общую нескладность фигуры многократно обыгрывали в шаржах. «Я всю жизнь работаю. Как вол! Как трактор!» - писал о себе Корней Иванович Чуковский. И это действительно было так, хотя его титаническая работоспособность для многих читателей детских стихов писателя, не знакомых с его многочисленными специальными литературоведческими статьями и переводами, оставалась скрытой. Как и Ханс Кристиан Андерсен, Чуковский многократно переделывал каждую свою строчку. «Никогда я не наблюдал, чтобы кому-нибудь другому с таким трудом давалась сама техника писания», - замечал он про себя.

Корней Иванович Чуковский
Из наших современников синдромом Марфана, возможно, страдал биолог Г. В. Никольский. Ко времени окончания Московского университета он имел уже пять печатных трудов. За 30 последующих лет работы число его печатных публикаций превысило 300, причем среди них было около десяти книг. Такой потрясающей работоспособностью может похвастаться далеко не каждый даже очень способный ученый!

Георгий Васильевич Никольский
Можно ли после этого утверждать, что любые обусловленные генами нарушения в развитии являются безусловно вредными?
skeletos.zharko.ru
В последние годы отмечается значительный рост наследственных заболеваний с поражением соединительной ткани. Некоторые специалисты считают, что причина такого явления кроется в накоплении в процессе эволюции человека новых генетических изменений (мутаций), к чему приводят внутренние и внешние факторы окружающей среды. Другие полагают, что на самом деле просто возросли диагностические возможности в связи с совершенствованием медико-генетических знаний, которые позволяют распознать генетическую патологию и поставить правильный диагноз.
Большую группу наследственных заболеваний составляет поражение соединительной ткани. Так как она является неотъемлемой частью всех органов и систем в организме, то такие патологии отличаются множеством нарушений и клинических симптомов. Одним из самых известных генетических заболеваний с поражением соединительной ткани считается синдром Марфана.
Такая патология характеризируется большой вариабельностью симптоматики (от скрытых форм до несовместимых с жизнью вариантов течения) и чаще всего включает поражения сердечно-сосудистой системы, глаз, центральной нервной системы и опорно-двигательного аппарата.
Встречается заболевание достаточно редко – 1 случай на 10 000 человек. Но если обратиться к статистике европейских стран, то частота значительно выше – 1-3 случая на 5000 человек, что связано с большей доступностью специфической диагностики скрытых форм патологии.

Известные люди тоже болели синдромом Марфана
Причины и генетика патологии
Впервые болезнь детально описал в 1896 году французский педиатр А.Марфан, в честь которого и назвали патологию. Он наблюдал за 5-летней девочкой астенического телосложения с непропорционально длинными конечностями и врожденной арахнодактилией (длинными пальцами). К середине 20-х годов прошлого столетия уже имелось множество подобных описанных клинических случаев у детей и взрослых. Американский генетик Мак Кьюсик провел детальное исследование мутаций хромосом и открыл новую группу наследственных заболеваний соединительной ткани, куда и отнесли синдром Марфана.
По этиологии синдром Марфана является генетическим заболеванием с аутосомно-доминантным типом наследования, с различной степенью экспрессивности (клинических проявлений генетических изменений). Примерно 85% случаев недуга носит наследственный характер, то есть наследуется от родителей. Остальные случаи являются новыми, то есть возникают вследствие новых спонтанных мутаций, а не передаются по наследству.
Непосредственная причина патологии в 95% случаев – это мутация в гене, которые кодирует строение фибриллина-1 и/или фибриллина-2. Локализируется мутация гена FBN1 и FBN2 в хромосоме 15 и 3.

Синдром Марфана наследуется по аутосомно-доминантному типу
Фибриллин – это основа эластических волокон соединительной ткани гликопротеиновой природы. Он составляет каркас межклеточного вещества, сосудистых стенок, хрящей, хрусталика глаза и многих других органов и тканей. В случае наличия описанной мутации у пациента соединительная ткань отличается повышенной способностью к растяжению, становится менее прочной и выносливой к механическим воздействиям, что и становится причиной клинических проявлений синдрома.
Примерно в 5% случаев непосредственной причиной синдрома Марфана (атипичные формы патологии) является точковая мутация гена, который кодирует строение α2-цепи коллагена первого типа.
Классификация
Согласно МКБ-10, синдром Марфана входит в класс врожденных дефектов развития и хромосомных патологий, тут патологию можно найти под шифром Q87.4.
Детальной клинической классификации недуга на сегодняшний день не существует, но выделяют несколько форм болезни, в зависимости от тех или иных критериев.
В зависимости от выраженности симптомов:
- стертая форма – признаки патологии мало выражены и могут оставаться незамеченными на протяжении всей жизни, как правило, изменения касаются не более 2 систем органов;
- клинически выраженная форма – симптомы патологии хорошо заметны и встречаются более чем в 2 системах органов.

Внешний вид ребенка, больного синдромом Марфана
В зависимости от генетического фактора:
- семейная форма диагностируется в случаях, когда болезнь передается по наследству;
- спорадическая форма определяется тогда, когда патология обусловлена новой спонтанной мутацией у индивида и при этом не встречается у его родственников.
Симптомы синдрома Марфана
Признаки синдрома Марфана очень разнообразны. Поэтому их рассматривают с точки зрения поражения отдельных органов и систем:
- опорно-двигательного аппарата;
- органов зрения;
- сердечно-сосудистой системы;
- нервной системы;
- дыхательной системы;
- кожных покровов и мягких тканей;
- прочих органов и систем.
Опорно-двигательный аппарат
Патологическая соединительная ткань обусловливает развитие ряда специфических фенотипических признаков и деформаций скелета у пациентов с данной патологией.

Характерный внешний вид пациентов с синдромом Марфана
Как правило, внешне пациенты с синдромом Марфана выглядят достаточно специфически. Для них характерно:
- астеническое телосложение;
- высокий рост;
- плохое развитие подкожной жировой клетчатки, из-за чего люди выглядят худощавыми;
- очень длинные верхние и нижние конечности при относительно коротком туловище;
- череп вытянутый (долихоцефалический);
- удлиненные пальцы – паукообразные (арахнодактилия);
- лицо узкое, вытянутое по вертикали;
- готическое верхнее небо;
- недоразвитие скул;
- выступающая нижняя челюсть (прогнатизм);
- неправильный рост (скученность) зубов и патологический прикус;
- гипермобильность суставов, их «разболтанность;
- глубоко посажены в черепе глаза.

Пример того, как определить наличие арахнодактилии, характерной для синдрома Марфана
По мере роста ребенка могут появляться различные деформации скелета. Чаще всего появляются искривления позвоночного столба. У пациентов диагностируют сколиоз, патологический кифоз и лордоз, осанку по типу «прямой спины» (сглаживание физиологического поясничного лордоза). Также появляются подвывихи и вывихи в шейном отделе позвоночника, примерно у 20% больных диагностируют поясничный спондилолистез.

Выраженная степень арахнодактилии при синдроме Марфана со специфическими контрактурами пальцев
Среди других скелетных деформаций встречается:
- протрузия вертлужной впадины тазобедренного сустава 2-3 степени, которая становится причиной диспластического коксартроза и инвалидности многих пациентов, если не выполнить операцию эндопротезирования;
- килевидная и вдавленная деформация грудной клетки;
- плоскостопие (продольное и поперечное).
Для пациентов в СМ также характерна остеопения (снижение минеральной плотности костей) и частые патологические переломы костей на ее фоне, а также склонность к привычным вывихам, например, плеча.

Вдавленная деформация грудной клетки у пациента с синдромом Марфана и результат ее хирургической коррекции
Сердечно-сосудистая система
Среди поражений кардиоваскулярной системы при СМ чаще всего встречаются:
- пролапс створок митрального клапана с регургитацией или без
- миксаматоз сердца;
- дилятационная кардиомиопатия с развитием сердечной недостаточности;
- аневризмы аорты и других сосудов (мозговых, почечных, пр.);
- расширение легочной артерии и различных отделов аорты.
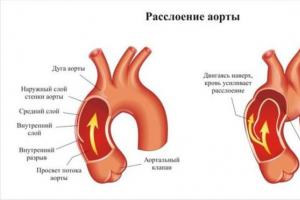
Именно кардиоваскулярные патологические изменения при СМ определяют прогноз и продолжительность жизни пациентов. Примерно 90% всех пациентов с данной генетической патологией умирают в возрасте 40-50 лет вследствие таких осложнений, как расслоение и разрыв аневризмы аорты, других сосудов, прогрессирующей недостаточности сердца вследствие дилатации его камер и изменений клапанного аппарата.
При наличии СМ возможно наличие врожденных пороков сердца у детей. Чаще всего встречаются коарктация аорты, стеноз (сужение) легочной артерии, дефект межжелудочковой и межпредсердной перегородки.
Также такие пациенты склонны к различным сердечным аритмиям, среди которых и опасные для жизни (мерцательная аритмия, желудочковая тахикардия и экстрасистолия), к инфекционному эндокардиту.

Аневризмы аорты и их разрыв чаще всего становятся причиной смерти пациента с синдромом Марфана
Орган зрения
Патологические изменения глаз являются весьма характерными для данного недуга. Примерно у 60-80% пациентов диагностируется дислокация хрусталика из-за слабости его связочного аппарата, причем еще в младенческом возрасте. Среди других характерных признаков:
- уплощение роговицы;
- увеличение размеров глазного яблока в длину;
- миопия или гиперметропия;
- нарушение процесса аккомодации из-за недоразвития цилиарной мышцы.
В случае выявления описанных поражений органа зрения у новорожденного ребенка следует задуматься о возможной генетической патологии.

Дислокация хрусталика – типичный признак синдрома Марфана
Нервная система
Из-за патологического строения стенок сосудов у пациентов с СМ повышен риск геморрагических инсультов, а также кровоизлияний в мозг при разрыве сосудистых аневризм, субарахноидальных кровотечений.
Среди аномалий развития встречается эктазия твердой мозговой оболочки . Чаще всего приходится сталкиваться с пояснично-крестцовой эктазией мозговой оболочки (выпячивание твердой мозговой оболочки за пределы позвоночного канала через дефект в строении позвонков). Это большой критерий СМ, который встречается в 40% случаев заболевания.
У части пациентов встречаются отклонения в интеллектуальном развитии, но большинство людей с СМ характеризируются высокими показателями IQ.
Органы системы дыхания
В большинстве случаев изменения бронхолегочного аппарата диагностируются случайно. Характерно развитие булл в верхних частях легких, которые иногда могут разрываться с развитием спонтанного пневмоторакса.
Также из-за деформаций грудной клетки пациенты склонны к развитию эмфиземы легких, частых инфекционных заболеваний органов дыхания и дыхательной недостаточности.

Буллезная эмфизема может быть причиной спонтанного пневмоторакса у пациентов с синдромом Марфана
Кожа и мягкие ткани
Встречается повышенная растяжимость кожного покрова, которая сочетается с развитием атрофических стрий. Последние появляются спонтанно, они никак не связаны с колебанием веса, беременностью или гормональными нарушениями. Подкожный жир выражен слабо у пациентов с синдромом Марфана. Они часто страдают рецидивирующими грыжами передней брюшной стенки.
Встречаются и многие другие патологические симптомы поражения прочих органов и тканей при СМ. Например, опущение почек (нефроптоз), выпадение мочевого пузыря и матки у женщин, варикозное расширение вен, хронические запоры и др.

Диагностика синдрома Марфана
Диагностика при синдроме Марфана носит в основном клинический характер. Обязательно учитывают анамнез, в том числе и семейный (наличие подобных проблем у кого-то из родственников), данные объективного обследования и осмотра. Также проводят множество дополнительных диагностических процедур для выявления патологии тех или иных органов и систем. Для этого применяют ЭКГ, УЗИ сердца и сосудов, рентгенографию органов грудной клетки, КТ, МРТ внутренних органов, позвоночника, головного мозга, офтальмоскопию и прочие исследования органа зрения, аортографию, ангиографию и много других методик, в зависимости от клинической ситуации и симптомов болезни.
Существуют общепринятые диагностические критерии синдрома Марфана (большие и малые), которые позволяют с большой степенью вероятности поставить правильный, но предварительный диагноз пациенту.

Окончательный диагноз синдрома Марфана выставляют только после анализа генотипа (ДНК-диагностика) и выявления специфической мутации в гене, ответственном за продукцию фибриллина, с помощью молекулярно-генетических методик.
Лечение заболевания
К сожалению, на сегодняшний день вылечить синдром Марфана, как и повлиять на его причину, невозможно. Терапия в основном направлена на улучшение качества жизни больного человека, устранение симптомов и профилактику осложнений.
Лечение синдрома Марфана должно быть комплексным и может включать как консервативные, так и хирургические методики.
Больные должны наблюдаться у различных докторов: кардиолога, окулиста, травматолога-ортопеда, клинического генетика, невролога и др.

Лечением синдрома Марфана должна заниматься целая команда специалистов
В случае выявления тех или иных кардиоваскулярных проблем назначают комплексное лечение, медикаменты для профилактики осложнений. Проводят медикаментозную коррекцию аритмий, частоты сердечных сокращений, артериального давления. Обязательно всем пациентам назначают прием бета-блокаторов, если нет противопоказаний к этой группе средств, которые имеют протекторный эффект в отношении возможного расслоения аорты.
В случае патологии клапанного аппарата сердца, аневризме аорты и других сосудов, расслоении их стенок может понадобиться операция. Также хирургическое вмешательство используют для коррекции деформаций опорно-двигательного аппарата.
В случае проблем с органом зрения также проводят оперативные вмешательства при эктопии хрусталика, лазерную коррекцию зрения, подбирают очки или контактные линзы.
В целом подбор лечебных методик и спектр применяемых средств очень индивидуальны. Это полностью зависит от присутствующих симптомов и степени выраженности болезни у конкретного пациента.

Основная задача в лечении пациентов с Синдромом Марфана заключается в предотвращении сердечно-сосудистых повреждений
Прогноз
Синдром Марфана отличается, как правило, хроническим прогрессирующим течением. Продолжительность жизни пациентов при условии полноценного комплекса лечебных мероприятий в среднем составляет 45 лет. Основными факторами риска преждевременной смерти выступают осложнения, которые возникают вследствие патологии кардиоваскулярной системы.
Синдром Марфана и беременность
Пациентки с СМ могут иметь детей, причем здоровых, но это очень опасно по двум причинам:
- Беременная женщина имеет очень высокий риск летальных осложнений СМ со стороны сердечно-сосудистой системы. Так как при вынашивании ребенка создается усиленная нагрузка на организм матери, а особенно на сердце и сосуды, то очень большие шансы у пациенток с синдромом Марфана получить разрыв аневризмы, ее формирование, расслоение аорты и прочие смертельно опасные осложнения.
- Риск передачи данной наследственной патологии своему ребенку составляет 50%.
Возможна ли профилактика болезни?
К сожалению, специфической профилактики синдрома Марфана не существует. Семейная пара, в которой один или оба родителя страдают данной патологией, в обязательном порядке должны планировать беременность и пройти медико-генетическое консультирование у врача-генетика. Вместе с этим можно проводить пренатальную (дородовую) диагностику на предмет наличия СМ у плода.
Существуют заболевания, причина которых скрыта в генетическом дефекте, а их проявления выражаются в сочетании определённых симптомов. Эти болезни ещё называют наследственными синдромами. Науке известно более 6000 генетических недугов, многие из которых встречаются крайне редко. Но некоторые заболевания известны врачу любой специальности, к таким болезням относится синдром Марфана.
Пациентов с этим диагнозом можно увидеть на приёме у разных специалистов, поскольку болезнь протекает с поражением многих органов и систем. Родителям необычных детей нужно знать, как проявляется этот недуг, и какие действия помогут улучшить качество жизни ребёнка.
Врач-педиатр, неонатолог

Синдром Марфана – генетический недуг, который выражается в недоразвитии соединительной ткани. Заболевание относится к редким, по статистике наследственная болезнь обнаруживается у 1 из 10000 детей. Возникновение синдрома не зависит от расовой принадлежности и пола малыша, и с одинаковой частотой проявляется, как у мальчиков, так и у девочек различных национальностей.
Историческая справка
Первые упоминания о необычном недуге можно обнаружить в трудах американского офтальмолога Э. Вильямса, который в 1875 году описал признаки идентичного смещения хрусталиков глаз у родных брата и сестры. Кроме офтальмологических проблем эти дети имели повышенную подвижность суставов и высокий рост.
Известность болезнь приобрела позже, через 20 лет, когда французский педиатр Антуан Марфан представил свои наблюдения за 5-тилетей больной. Маленькая пациентка отличалась необычными аномалиями скелета и быстрым прогрессированием недуга. Синдром был назван в честь французского доктора, хотя впоследствии стало известно, что наблюдаемая им девочка страдала другой наследственной патологией – врождённой контрактурной арахнодактилией.
Можно найти множество примеров обнаружения синдрома у талантливых, знаменитых людей. Считается, что этим недугом страдали скрипач Никколо Паганини, американский президент Авраам Линкольн, русский композитор Сергей Рахманинов и другие известные личности. Некоторые исследователи считают, что неординарность людей с синдромом Марфана объясняется увеличенной концентрацией адреналина в крови. Этот гормон вызывает повышение активности и развитие незаурядных способностей.
Почему проявляется генетический синдром?
Причиной развития недуга считается мутация в гене FBN1, который располагается в 15 хромосоме и отвечает за нормальное производство фибриллина 1. Этот белков соединительной ткани является одним из главных компонентов, придающих ей эластичность и способность к сокращению.
Первыми при генетическом синдроме поражаются структуры, содержащие наибольшее количество важного белка – стенки кровеносных сосудов, связочный аппарат, цинновая связка глаза. Изменённая соединительная ткань не способна выполнять своей функции, выдерживать физическую нагрузку в связи с потерей прочности и упругости, у ребёнка возникают симптомы заболевания.
Недуг относится к генетическим и передаётся от родителей по аутосомно-доминантному типу. Риск появления малыша с наследственным синдромом очень высокий, если у мамы или папы имеются признаки болезни. В 75% случаях заболеваний прослеживается появление недуга в каждом поколении семьи. У 25% больных определяется новая, спонтанная мутация, не находится чёткой связи с наследованием.
Соединительная ткань не образует отдельного органа в человеческом теле. Но её клетки располагаются во всём организме. По средствам этих структур выполняются опорная, защитная и трофическая функции, образуется своеобразный каркас и покровы всех органов. К разновидностям соединительной ткани относят хрящевую, костную, мышечную, жировую ткани, кровь и лимфу. Поэтому системные заболевания, связанные с тканевой патологией, отличаются большим многообразием проявлений.

Классификация синдрома
Болезнь отличается многообразием проявлением и различной их выраженностью. Этот недуг может быть длительное время нераспознанным, а некоторые отличительные особенности ребёнка расцениваться, как вариант нормы. В то же время существую формы, при которых характерные признаки болезни видны уже в роддоме.
Неонатальный вариант течения синдрома отличается выраженными признаками болезни у новорождённого, быстрым прогрессированием и высокой смертностью детей.
В зависимости от выраженности клинических проявлений различают 2 формы болезни:
- стёртую.
Признаки поражения органов незначительные и охватывают 1 – 2 системы организма;
- выраженную.
Данную форму определяют, если хотя бы одна из систем организма имеет серьёзные нарушения функций. Врач может поставить этот диагноз и в случае обнаружения умеренных поражений 2 – 3 систем организма.
Большое значение в определении прогноза заболевание имеет динамика нарушений (специалисты различают прогрессирующий и стабильный варианты синдрома).
Классические симптомы заболевания
Некоторые проявления наследственной болезни можно обнаружить даже у новорождённых малышей, такие крохи имеют большую длину тела, чем их сверстники, длинные пальцы, умеренные поражения костной системы и внутренних органов. Но характерная симптоматика формируется к 7 – 8 годам жизни ребёнка, со временем признаки болезни становятся более выраженными, возникают новые проявления. Все больные синдромом Марфана имеют схожие поражения внутренних органов, кроме того их внешний вид так же типичный.

Синдром Марфана проявляется у разных людей по-разному, это зависит от типа мутации в гене FBN1. Симптомы могут быть лёгкие, едва заметные, которые тяжело отличить от индивидуальных особенностей ребёнка. В других случаях наблюдается выраженная, классическая картина заболевания с развитием и прогрессированием главных симптомов.
Особенности внешности
Дети с патологией соединительной ткани значительно выше своих сверстников, они отличаются худощавым телосложением и непропорционально длинными и тонкими конечностями (долихостеномиелия). Пальцы на руках и ногах удлинённые, их ещё называют «паучьи», а размах рук превышает рост ребёнка. Тонкая, бархатистая кожа со слабо развитой подкожной жировой клетчаткой, склонна к образованию растяжек, стрий.
При рассмотрении лица больного можно заметить вытянутый овал, небольшую нижнюю челюсть, близко посаженные глаза. Возможен неправильный рост зубов, нарушенный прикус, а нёбо крохи расположено выше, чем у других детей.
Дети с синдромом Марфана отличаются живым характером, энергичностью, гиперактивностью. Среди этих ребят много талантливых, незаурядных личностей.

Патология опорно-двигательного аппарата
Поражение соединительной ткани значительно сказывается на строении и функционировании костной системы. Позвоночник ребёнка не способен выполнять свою опорную функцию, выдерживать возрастающую с ростом малыша нагрузку на него.
Возникают различные деформации позвоночного столба – сколиоз, кифоз, их сочетание. Из-за нестабильности связочного аппарата возникают подвывихи и вывихи шейного отдела позвоночника.
Грудная клетка ребёнка также деформируется, возникают её смещение наружу (килевидная грудь) или наоборот, западение грудины (воронкообразная, впалая грудь). Также у больных детей нередко развиваются плоскостопие, рекурвируются колени (избыточно разгибаются в суставах).
Кроме того, у ребят наблюдается повышенная гибкость, гипермобильность суставов, что связано с растяжимостью хрящевой ткани, связок и суставов. Такие дети отличаются большей пластичностью, чем их сверстники. Нередко родители радуются этой особенности малыша, особенно если деформации скелета выражены незначительно, и решают отдать кроху в спортивную секцию.
Занятия спортом опасны для детей с поражением соединительной ткани. Хотя анатомические структуры у этих малышей обладают большей растяжимостью, из-за недостаточной прочности тканей нередко возникают травмы и разрывы сухожилий, суставных сумок.

Патология системы кровообращения
Одним из ведущих признаков, определяющих течение и исход заболевания, является поражение сердца и сосудов. Дефект соединительной ткани проявляется патологией строения стенок крупных сосудов, клапанов и перегородок сердца.
В тяжёлых случаях малыш рождается с врождённым пороком сердца, угрожающим жизни и требующим незамедлительной хирургической коррекции. Нередко у детей выявляются признаки пролапса клапанов, чаще митрального и аортального. Из-за недостаточной эластичности соединительной ткани клапаны (своеобразные створки, которые препятствуют обратному току крови) не могут выполнить свою функцию. Возникают нарушения кровообращения, которые проявляются обмороками, головокружением, одышкой, повышенной утомляемостью.
Самым опасным состоянием при синдроме Марфана считается расширение, расслаивание стенки и разрыв аорты. Её поражение носит врождённый характер и имеет тенденцию к прогрессированию, принося всё новые опасные патологические симптомы. Поэтому наблюдение за состоянием сердечно-сосудистой системы и своевременное предотвращение и лечение осложнений – главная задача в терапии синдрома Марфана.
Аорта – самый крупный непарный сосуд, который разносит обогащённую кислородом кровь по всему организму. Аневризмой называют участок расширения стенок сосуда, который обусловлен слабостью стенок артерии. Опасным осложнением патологии считается расслоение стенки сосуда и пропитывание кровью поражённого участка. Прорыв артерии приводит к кровотечению, острой сердечно-сосудистой недостаточности, летальному исходу.
Другие патологии сердца при синдроме Марфана проявляются нарушением ритма и проводимости, развитием бактериальных осложнений – инфекционного эндокардита.
Поражение органов зрения
Около половины больных наследственным синдромом людей имеют эктопию хрусталика, которая развивается обычно до 4-хлетнего возраста и со временем прогрессирует. В норме хрусталик удерживается в правильном положении с помощью цинновых связок. У детей с патологией соединительной ткани возникает слабость связочного аппарата глаза, что приводит к развитию заболеваний.
Эктопия хрусталика – изменение положения биологической линзы по типу подвывиха (частичное смещение) или вывиха (выпадение хрусталика в переднюю камеру глаза или стекловидное тело).
Кроме того со стороны органов зрения возникают такие патологии, как близорукость, повышение внутриглазного давления – глаукома, отслойка сетчатки, колобома радужки и другие заболевания.

Изменения в других органах и системах
Поскольку соединительная ткань является составной частью любого органа, у ребёнка могут возникнуть разнообразные патологии. Со стороны нервной системы иногда возникает такая патология, как менингоцеле. Из-за врождённого дефекта позвоночника происходит выпячивание спинного мозга и его оболочек, обычно это наблюдается в пояснично-крестцовой области.
Дети с синдромом Марфана имеют слабо развитую мышечную ткань. Нередко у них возникают грыжевые выпячивания, которые рецидивируют, возникают снова даже после оперативного лечения. Часто наблюдается изменение положения внутренних органов – опущение почек, матки, мочевого пузыря.
Даже небольшие травмы и падения опасны для больных генетическим синдромом. Вывихи, разрывы связок, которые долго не заживают, часто приносят беспокойства малышу. Со стороны дыхательной системы отмечаются врождённые пороки развития лёгких, кистозные изменения ткани, спонтанные пневмотораксы (надрыв плевры и скопление воздуха в плевральной полости).
Синдром Марфана развивается в результате мутации в определённом гене, который кодирует синтез фибриллина 1. В настоящее время выделено более 100 различных дефектов в генетическом материале, ответственном за синтез фибриллинов 1, 2, 3. Эти патологии имеют схожие проявления, но не являются синдромом Марфана. В некоторых случаях говорят о фибриллинопатиях, нарушении синтеза белков соединительной ткани.
Диагностика синдрома Марфана
Для правильной постановки диагноза малышу придётся пройти комплексное обследование, получить консультацию у многих специалистов.

Определение анамнеза
Поскольку патология имеет наследственную природу, в большинстве случаев удаётся проследить заболевание в семье. Нужно помнить, что недуг может протекать в различных формах и многие люди, с лёгким течение синдрома, не догадываются о своём заболевании на протяжении всей жизни.
Особенность клинических проявлений
Поскольку болезней, связанных с патологией соединительной ткани, существует множество, иногда бывает сложно правильно поставить диагноз ребёнку. Чтобы справится с этой задачей в 1996 году генетиками и клиницистами были разработаны современные критерии, с помощью которых можно определить синдром Марфана. В основу диагностики были положены «большие» и «малые» признаки заболевания. Их сочетание оценивается специалистом и решается вопрос о наличии генетического синдрома.
- большие критерии.
К ним относятся:
- увеличение роста в большей степени за счёт верхней части тела;
- грубая деформация грудной клетки и позвоночника;
- продольное плоскостопие;
- невозможность полностью разогнуть конечность в коленных и локтевых суставах (контрактуры);
- эктопия хрусталика;
- расширение и расслоение восходящей части аорты и другие признаки.
Некоторые из этих симптомов можно встретить у абсолютно здоровых детей. В диагностике генетического синдрома большую роль играет именно их сочетание. Триада Марфана включает в себя патологию костно-суставной системы, органические изменения в сердце или крупных сосудах, болезни глаз;
- малые критерии.
Эти признаки в меньшей степени указывают на наследственный дефект, но их наличие и сочетание с большими критериями подтверждает диагноз синдром Марфана.

К ним относятся:
- высокая подвижность суставов;
- аномалии зубов, нёба;
- гипоплазия радужной оболочки глаза, цилиарной мышцы, увеличение длины глазного яблока;
- пролапс митрального клапана;
- патологии бронхо-лёгочной системы, спонтанный пневмоторакс и другие нарушения.
Для уточнения внешних изменений у ребёнка используются различные способы: измерение роста, длины кисти, соотношение размеров верхней части туловища к нижней и другие. Особо показательны в диагностике синдрома следующие исследования:
- тест запястья.
Врач просит ребёнка обхватить запястье одной руки большим пальцем и мизинцем другой руки, образуя «браслет». В пользу наследственного недуга говорит лёгкое смыкание кисти на запястье другой руки, нахождение фаланг мизинца и большого пальца друг на друга;
- тест большого пальца.
Исследователь просит малыша попытаться дотянутся большим пальцем до предплечья этой же руки. Тест считается положительным, если ногтевая фаланга пальца ребёнок с лёгкостью достаёт до лучевой кости предплечья.

Лабораторные исследования
Обычные клинические и биохимические анализы крови и мочи не показательны при синдроме Марфана, изменений в них может не быть. Помочь в установлении диагноза поможет обнаружение продуктов метаболизма соединительной ткани в моче.
Резкое увеличение оксипролина и гликозаминогликанов в суточной моче может говорить о развитии осложнений у ребёнка (прогрессировании сердечной недостаточности, отслойке плаценты, развитии пневмоторакса, пневмонии).
С помощью современных методов исследования, молекулярно-генетической диагностики можно обнаружить характерную для данного генетического синдрома мутацию в гене FBN1.
Такое заболевание, как синдром Билса отличается от синдрома Марфана дефектом синтеза другого белка – фибриллина 2, но клинические симптомы этих недугов схожи. Различить их можно только с помощью молеклярно-генетической диагностики и выявления особого симптома «мятого уха», что указывает на наличие синдрома Билса.

Инструментальные методы
Специфические проявления заболевания можно обнаружить во многих органах, поэтому заподозрив у ребёнка наследственный синдром, проводятся различные исследования. Патологию костно-суставной системы обнаруживают с помощью рентгенографии и компьютерной томографии.
Болезни сердца и сосудов диагностируются благодаря ЭКГ и ЭхоКС, МРТ. С помощью УЗИ обнаруживаются патологии внутренних органов, смещение их положения в брюшной полости. В исследовании органа зрения помогут такие методы, как офтальмоскопия, биомикроскопия.
Консультация специалистов
Ребёнок с наследственным синдромом состоит на учёте у многих врачей: генетика, травматолога, хирурга, офтальмолога, кардиолога и других специалистов.
Диагностика синдрома Марфана у детей младшего и среднего возраста затруднена. Это объясняется быстрым ростом ребёнка и изменением нарушений. Выраженность некоторых патологий при правильном лечении уменьшается, в то же время при отсутствии необходимой терапии клинические проявления становятся более яркими, появляются новые симптомы. Поэтому стоит сохранять настороженность в отношении детей с подозрением на генетический синдром, регулярно обследовать и лечить малыша.
Лечение синдрома Марфана
Специфической терапии, направленной на устранение причины заболевания не существует. В настоящее время не разработаны методы влияния на наследственный аппарат клетки. Поэтому основная цель лечения синдрома Марфана – предотвращение прогрессирования заболевания, борьба с симптомами болезни:
- патология сердца и сосудов.
Самым опасным проявлением недуга считается аневризма, расширение участка аорты. Коварство болезни заключается в непрерывном, длительном прогрессировании симптомов. Случается, что опасный симптом формируется к 18 годам, поэтому важно уделять достаточно внимания ежегодному обследованию и лечению сердечно-сосудистой системы.
Грубые пороки развития сердца и сосудов, тяжёлые осложнения хронических болезней лечатся оперативно. Из лекарственных препаратов назначаются ингибиторы АПФ, блокаторы кальциевых каналов. Применение b-адреноблокаторов (пропанолола, атенолола) показано при расширении корня аорты, пролапсе клапанов, аритмиях.
Назначение препаратов и подбор необходимой дозировки должно проводиться врачом-кардиологом с учётом данных обследования ребёнка. Необоснованное назначение лекарственных средств может привести к ухудшению состояния ребёнка;

- болезни опорно-двигательного аппарата.
Существую исследовании, указывающие на дефицит некоторых макроэлементов (кальций, цинк, кобальт, магний) и белков, необходимых для строительства соединительной ткани при синдроме Марфана. Поэтому для предотвращения прогрессирования патологии назначаются витаминно-минеральные комплексы, гиалуроновая кислота, викасол, колекальциферол. Оперативное лечение показано при грубых патологиях развития скелета;
- заболевания органов зрения.
Исправление патологии зрения проводится с помощью подбора специальных очков, контактных линз, оперативного лечения катаракты и глаукомы, смещения хрусталика.
Опасным осложнением синдрома является отслойка сетчатки. Эта патологии возникает при активном занятии спортом у ребят с дефектом соединительной ткани. Повышенная физическая нагрузка, прыжки, травмы приводят к отделению тонкой сетчатой оболочки от сосудистой. Такое нарушение сопровождается резким снижением остроты зрения, которое не всегда является обратимым. Поэтому ребятам с генетическим синдромом следует избегать чересчур активных занятий, для таких детей хорошо подходит плавание в бассейне;
- нарушенный обмен веществ.
Для улучшения метаболизма рекомендовано использовать в комплексном лечении аскорбиновую и янтарную кислоты, карнитин, препараты магния, токоферола ацетата. С целью нормализации обмена хрящевой ткани используются глюкозаминсульфат, хондроитинсульфат.
Существуют данные, указывающие на необходимость введения диеты с высоким содержанием магния для детей с синдромом Марфана. Этот элемент помогает справиться с повышенным содержанием катехоламинов в крови и способствует восстановлению дефектов кровеносных сосудов. Хорошо включать в ежедневный рацион орехи, какао, гречневую и ячневую каши, сухофрукты.
Прогноз и профилактика
Течение болезни во многом зависит от выраженности клинических проявлений и качества проведённого лечения. Особую опасность для жизни ребёнка представляют пороки развития сердца и сосудов, их патологические изменения, поэтому терапии этой группы заболеваний отводится особое место.
Родителям девочек нужно знать об опасности будущей беременности для здоровья больной наследственным синдромом. Именно во время вынашивания ребёнка увеличивается риск развития и расслаивания аневризмы аорты. Это связано с повышенной нагрузкой на систему кровообращения и гормональными изменениями.
В целом, при правильном лечении и вовремя оказанной помощи пациенты с генетическим синдромом доживают до глубокой старости. Болезнь привносит ограничения в выборе будущей профессии. Таким детям лучше искать занятие не связанное с повышенной физической нагрузкой, поднятием тяжестей.
Профилактика недуга заключается в своевременном диагностировании синдрома в семье и медико-генетическом консультировании будущих родителей.

Приветствую, дорогие читатели. Сегодня хотел бы поговорить про высоких людей с Синдромом Марфана, для которых высокий рост – это не просто особенность организма, а симптом серьезной болезни, требующей к себе постоянного внимания.
— заболевание наследственного типа, при котором поражается соединительная ткань с вовлечением в процесс скелетно-мышечной системы и глаз. Установлено, что причиной патологии является мутация гена фибриллина FBN1. Заболевание полиморфно — может протекать с разной выраженностью клинической картины, и характеризуется появлением все новых типов мутации в генах.
Синдром Марфана получил своё название от фамилии французского педиатра А. Марфана, который вперые представил описание 5-летней девочки Габриель с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя.
Распространенность синдрома — 1 случай на 10000 человек. Риск рождения ребенка с синдромом Марфана повышается после достижения отцом возраста 35 лет и достигает 50% при наличии патологии у одного из родителей. Врожденная аномалия наследуется по аутосомно-доминантному типу. В ее основе лежит дефект важнейшего гена, отвечающего за синтез коллагена.

Во время внутриутробного развития происходит нарушение формирования волокон соединительной ткани, утеря ими прочности, в результате чего волокна не способны выдерживать естественные нагрузки. Поэтому наибольшие атипичные изменения претерпевают крупные сосуды, клапаны сердца, связки глаза, твердое небо, скелет и мышцы.
Без адекватной терапии продолжительность жизни людей с синдромом Марфана не более 40 лет. Терапия позволяет увеличить этот срок вдвое и более.
История заболевания
В 1876 г. симптомы неизвестной патологии были отмечены доктором Вильямсом, но клинические наблюдения проводились гораздо позже — в 1896 г. педиатром из Франции А. Марфаном. Врач в течение 5-ти лет оценивал состояние девочки с неизученными ранее аномалиями, заключающимися в прогрессировании дистрофии скелета и мышечной ткани.
К середине 20-го века имелось множество описанных случаев, когда у больных наблюдались симптомы, близкие к патологии Марфана, и все они относились к заболеваниям наследственного типа. Среди таких случаев — расслоение аорты, пороки сердца, эктопия хрусталиков, сопровождающиеся деформацией костей (грудной клетки, позвоночника) и внешними отклонениями от нормы (высокий рост, худоба, длинные конечности). Американским генетиком МакКьюсиком было проведено детальное исследование мутаций хромосом и открыта новая группа заболеваний соединительной ткани.

Симптомы синдрома Марфана
Многообразие вариантов генетической мутации обуславливает различные формы течения болезни. Нередко они малозаметны, иногда приводят к инвалидизации человека в раннем возрасте. Частый признак синдрома Марфана — высокий рост (до 200 см.), при этом туловище непропорционально короткое, а конечности удлиненные и тонкие. Пальцы у больных длинные, паукообразные (арахнодактилия) Из-за недоразвития подкожной клетчатки и мышечной дистрофии страдающие синдромом Марфана имеют астеническое телосложение.
Прочие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
— гиперподвижность суставов;
— аномалии строения тазобедренного сустава;
— кифоз, сколиоз;
— вывихи шейного сегмента позвоночника;
— деформация грудной клетки;
— плоскостопие;
— глубокая посадка глаз;
— уменьшенная нижняя челюсть, нарушение роста зубов;
— высокое нёбо;
— атрофические «растяжки» на коже;
— паховые грыжи, частые разрывы связок.
Более серьезные изменения при синдроме Марфана протекают в организме. Самые тяжелые из них развиваются со стороны сердца и сосудов и могут привести к смерти ребенка еще на первом году жизни. Среди них:
— дефекты ветвей легочной артерии, аорты (расширения, аневризмы, расслоения);
— пороки сердца (чаще — поражения клапанов);
— стенозы артерий.
Подобные нарушения вызывают тахикардию, мерцательную аритмию вплоть до фибрилляции предсердий или развития сердечной недостаточности.
Со стороны глаз наблюдается выраженная миопия, вывих хрусталика, аномалии развития роговицы, уменьшение в размерах радужки, косоглазие, патологии сосудистой стенки сетчатки. При прогрессирующем вывихе хрусталика или при отслойке сетчатки уже в раннем возрасте больные могут полностью потерять зрение.
Со стороны нервной системы
при синдроме Марфана происходит растяжение твердой мозговой оболочки и выбухание ликвора в костные дефекты в пояснично-крестцовом отделе позвоночника (дуральная эктазия). Легкие страдают гораздо реже, так как незначительные нарушения их работы не оказывают влияния на дыхательную функцию. Но в отдельных случаях снижение эластичности альвеол может привести к спонтанному пневмотораксу, развитию дыхательной недостаточности. Прочими симптомами патологии могут быть
эктопия почек, деформации мочевого пузыря, половых органов.
Лечение и профилактика осложнений
Специфической терапии заболевания не существует: изменить гены еще до рождения ребенка невозможно. Лечение только симптоматическое и зависит от тех изменений в организме, которые развиваются у больного синдромом Марфана . Некоторые осложнения патологии можно успешно корректировать, другие — устранять оперативным путем.

Пациент должен наблюдаться у группы специалистов — офтальмолога, невролога, кардиолога, ортопеда, хирурга. Основное направление терапии — поддержка функций сердца и сосудов.
Методы лечения
:
— прием препаратов
(адреноблокаторы, антиаритмические лекарства, антикоагулянты и т.д.);
— хирургия пороков сердца
(дисфункции клапанов, расширения, расслоения легочной артерии), аорты, протезирование клапанного аппарата.
Нормализация зрения проводится при помощи коррекции миопии (ношение очков, линз), лечения катаракты, глаукомы, имплантации искусственного хрусталика.
При поражении суставов и позвоночника проводится оперативное лечение (протезирование, пластика суставов, устранение межпозвоночных грыж), выправление кифоза, сколиоза при помощи тракции, мануальной терапии. Из медикаментозных средств используются миорелаксанты, витамины группы В. Также применяется физиолечение, занятия ЛФК.
При поражении легких часто требуется хирургическое вмешательство (дренирование их полости).
Беременность больными синдромом Марфана должна строго планироваться и развиваться под контролем группы врачей, специализирующихся на лечении людей с подобными патологиями. Родоразрешение — только при помощи кесарева сечения. Еще до наступления беременности желательно обследоваться на предмет возможного прогрессирования расслойки аорты и, по возможности, провести операцию по замене части сосуда. Консультация генетика позволит рассчитать примерный риск по передаче заболевания по наследству.
Хоть синдром Марфана – очень редкое заболевание, есть немало знаменитостей, больных синдромом Марфан: Фло Хайман (призер Олимпийских игр по волейболу), Джон Тавенер (композитор), Джоуи Рамон (музыкант), Лесли Хорнби (фотомодель и певица) и другие.
Среди исторических личностей, известных во всем мире, с синдромом Марфана можно выделить:

Музыкант-скрипач Никколо Паганини . Поскольку Паганини умер еще до описания синдрома, данные о его заболевании исследуются по сохранившимся изображениям и дневнику лечащего врача. Скрипач имел характерную деформацию пальцев, высокий рост и худобу, непропорциональное развитие конечностей, впалую грудь, мышечную слабость.
Писатель Ганс Кристиан Андерсен . Имел угловатое лицо, был очень худым и длинноруким, рано заполучил проблемы со зрением.
Президент Америки Авраам Линкольн . Кроме всех внешних признаков синдрома Марфана у Линкольна наблюдались ревматические боли, «разболтанность» суставов, но, в то же время — хорошая физическая выносливость.
Писатель Корней Чуковский . Наличие большого непропорционального носа, длинных конечностей не помешало Чуковскому стать одним из лучших творцов современности и доктором филологии.
Осама Бен Ладен
. «Террорист №1» мира имел высокий рост и малый вес и большие проблемы с суставами и позвоночником, а также вытянутый череп и слишком узкое лицо.
Источники и ссылки:
1. — статья, в которой рассмотрены основные причины высокого роста человека.
2. Синдром Марфана в википедии — о синдроме Марфана в свободной энциклопедии.
3. Русскоговорящее сообщество людей с синдромом Марфана — форум о синдроме Марфана, общение, обсуждение болезни.
4. Международный фонд синдрома Марфана — фонд помощи больным с синдромом Марфана.
Синдром Марфана – это наследственное заболевание, передающееся по аутосомно-доминантному типу и характеризуется поражением соединительной ткани и ее компонентов.
Болезнь Марфана вызывается мутированием гена, кодирующего фибриллин -1.
Люди с синдромом Марфана имеют удлиненные конечности, паукообразные пальцы и слабый (недоразвитый) подкожно-жировой слой и сверхгибкие суставы (см. фото ниже).
Кроме изменений костно-суставной системы, характерны изменения зрительного анализатора и сердечно-сосудистой системы. Также возможно поражение нервной, дыхательной и других систем.
Впервые описал данную патологию Вильямс, который заметил у своих брата и сестры – выпадение хрусталика, при этом они были очень высокими и имели гипермобильные суставы. Затем заметил Марфан, врач – невролог, у которого в течение 20 лет наблюдалась женщина с подобными симптомами, а затем еще 20 детей.
Причины возникновения

Болезнь Марфана у детей наследуется по аутосомно-доминантному типу (т. е передается от родителя к ребенку).
Также возможны мутации за счет воздействия на организм женщины факторов внешней среды (ионизирующее излучение, лучевая терапия, радиация).
Причины возникновения и механизм развития заболевания недостаточно изучены.
Особая роль отводится нарушению процессов обмена, в результате которых накапливается в коллагеновых и эластических волокнах большое количество мукополисахаридов.
Это ведет к тому, что соединительная ткань перерастягивается, легко подвергается механическому воздействию и приводит к развитию клинической симптоматики.
Классификация
Выделяют следующие формы болезни Марфана:
В зависимости от генной предрасположенности:
- семейная (патология передается от родителя к ребенку);
- спорадическая (патология вызвана внезапным мутированием в геноме).
В зависимости от проявлений клиники:
- стертая, когда признаки заболевания практически не проявляются и могут быть не замечены в течение всей жизни. Патологические изменения выявляются в одной или двух системах.
- выраженная, когда признаки заболевания касаются двух и более органов и систем (сердце, кости и суставы, легкие, кожа, глаза).
Симптомы болезни Марфана
Синдромом Марфана у людей ведет к их выделению в обществе своим непропорциональным строением скелета. Для новорожденных на ранней стадии болезни, характерны длинные пальцы на руках, а к 7-9 годам у детей формируется развернутая клиническая картина.
У взрослых характерна различная симптоматика в зависимости от системы поражения:
- Нервная система: болезненность в поясничной области, головные боли,поражение симпатической и парасимпатической иннервации органов брюшной полости и малого таза (слабость кишечной стенки, недержание мочевого пузыря у ребенка). Также высок риск развития , субарахноидального кровоизлияния и разрыва аневризм головного мозга.
- Сердечно – сосудистая система: \пороки сердца (сужение легочной артерии, пролабирование створок двустворчатого клапана, дилатационная кардиомиопатия, расширение границ сердца (аорты и всех ее отделов), дефекты МЖП и МЖЖ перегородок. У больных может развиваться нарушение ритма и проводимости в виде аритмий.
- Опорно-двигательный аппарат: телосложение астенической формы (дети худые), высокий рост у мужчин 190±10 см, у женщин 179±8 см, слаборазвитый подкожно-жировой слой, длинные пальцы (паукообразные), череп и лицо вытянутые и узкие, недоразвитость скул, нарушение развития зубов и прикуса, вытянутая нижняя челюсть, готическое верхнее небо, гипермобильность суставов (смотрите на картинки выше). С возрастом ребенка может прогрессировать деформация позвоночного столба, с развитием . Также может деформироваться грудная клетка, образуется вдавление – «грудь сапожника». Деформированный тазобедренный сустав нередко приводит к инвалидности при неоказанном своевременном лечении.
- Орган зрения: смещение хрусталика за счет слабого связочного аппарата) на ранней стадии, уплощение роговицы, развитие близорукости или дальнозоркости, спазм аккомодации, отслоение сетчатки.
- Кожа и мягкие ткани: перерастяжение кожи с образованием стрий атрофического характера. Они возникают внезапно, не связаны с колебание веса людей, беременностью и гормональным фоном. Кожа липкая, потная, с мраморным оттенком. Подкожно-жировой слой слабо развит, поэтому у больных наблюдаются грыжевые выпячивания в области передней брюшной стенки.
- Дыхательная система: развитие буллезной , проявляющейся кашлем, одышкой, развитием дыхательной недостаточности и спонтанного .
Другие признаки :
- развитие опущения почек ();
- выпадение органов малого таза ( , или ее полное выпадение);
- запоры.
Диагностика
Диагностика основана на тщательном сборе анамнеза заболевания, выраженности клинической картины, данных осмотра, на результатах лабораторных и инструментальных методов исследований.
Сбор анамнеза включает в себя: наличие в семье данной патологии (родители, братья, сестры) или наличие факторов, провоцирующих мутацию в геноме человека.
К лабораторным методам относят: анализ генотипа ДНК с мутирующим геном, определение гликозаминогликанов в моче.
К инструментальным методам исследования относят:
- ЭКГ, служит для обнаружения патологии сосудов и сердца (ССС). Выявляют характерные нарушения ритма и проводимости в виде , желудочковой экстрасистолии, развитие дилатационной гипертрофии миокарда левого желудочка.
- ЭхоКГ, также служит для обнаружения патологии ССС. Выявляют расширение аорты и ее структур, пролабирование двустворчатого клапана, увеличение размеров левой половины сердца.
- проводится для определения осложнений (расслаивающаяся аневризма).
- Рентген органов грудной клетки (изменения скелета, расширение полостей сердца, корней легких и др.)
- Компьютерная томография, магнитно-резонансно-ядерная томография позволяет выявить патологии костно-суставной, нервной системы, нарушение кровообращения в сосудах головного и спинного мозга.
Данные методы исследования служат для обнаружения критериев синдрома Марфана в различных органах и системах. Они играют самую важную роль для постановки и подтверждения диагноза, а в последующем, для определения тактики лечения.
Существуют следующие критерии диагностики синдрома Марфана:
| Система | Большие критерии | Малые критерии |
| Опорно- двигательный аппарат Должны быть: 4 больших критерия, либо 2 больших и 1 малый. |
|
|
| Орган зрения | Смещение хрусталика | Уплощенная роговица, близорукость, дальнозоркость, недоразвитие радужки и цилиарной мышцы глаз. |
| Сердечно-сосудистая система | Расширение аорты и ее структур | Пролабирование двустворчатого клапана, расширение клапана легочной артерии у лиц, не достигших 40 лет, отложение солей кальция на створках двустворчатого клапана, расслаивание аорты. |
| Дыхательная система | Отсутствуют | Внезапно развивающийся пневмоторакс (скопление воздуха в грудной клетке), верхушечные буллы. |
| Кожа | Отсутствуют | Повторное развитие грыжевых выпячиваний, атрофические стрии. |
| Нервная система | Расширения сосудов оболочек спинного мозга в поясничном\крестцовом отделе позвоночного столба. | Отсутствуют |
| Генетические изменения | Наличие данных критериев у родителей, детей, братьев, сестер, бабушек, дедушек. Наличие мутирующего гена, кодирующего фибриллин 1. | Отсутствуют |
Для постановки диагноза «Синдром Марфана» учитывается один признак из перечня больших критериев или малый критерий, характерный каждой из пораженной систем, кроме опорно-двигательного аппарата, (необходимо, как минимум, 4 критерия), а также наличие в семейном анамнезе больных с данной патологией.
Лечение синдрома Марфана
От синдрома Марфана полностью избавиться и устранить механизм его развития невозможно. Лечение базируется на улучшении общего состояния больного, устранении клинических проявлений и проведении профилактических мероприятий, препятствующих развитию осложнений.
Следует обходить стороной и повседневные нагрузки, при которых возможно повышение внутригрудного давления, ведущему к развитию пневмоторакса (например, подъем тяжестей, подъем по этажам).
Больные с синдромом Марфана должны консультироваться у разных специалистов, в зависимости от клинически пораженных систем органов. Следует проходить медицинские осмотры каждые полгода в течение всей жизни.
Медикаментозное лечение

Медикаментозная терапия направлена на устранение клинической картины заболевания.
Со стороны сердечно-сосудистой системы рекомендованы β-адреноблокаторы (например: Анаприлин ), которые снижают скорость распространения пульсовых волн при стремительно растущем расширении аорты и обратного тока крови на двустворчатом клапане или клапане аорты.
β-адреноблокаторы также оказывают положительный эффект при нарушении ритма и проводимости, в сочетании с сердечными гликозидами.
Но следует помнить о существующих противопоказаниях данных групп препаратов:
- хронический обструктивный бронхит;
- снижение ;
- низкое артериальное давление.
Блокаторы каналов кальция используются при наличии противопоказаний к В-адреноблокаторам.
Хирургия
Хирургическое лечение проводится, если есть осложнения со стороны сердечно-сосудистой системы, с целью коррекции пораженных участков. Его проводят при пролабировании двустворчатого клапана и расслаивании аорты.
При этом осуществляется протезирование двустворчатого клапана.
У беременных с тяжелым течением болезни Марфана, роды разрешаются хирургическим путем.
Профилактика
С профилактической целью, во избежание развития инфекционных осложнений, образования тромбов и тромбоэмболии, назначаются антикоагулянты (гепарин), антибактериальная терапия и витаминотерапия.
- При синдроме Марфана с тяжелым поражением зрительного анализатора проводится хирургическая коррекция зрения, после которой пациенты должны носить очки или контактные линзы.
- Если возникают осложнения, проводят лазерную коррекцию глаукомы, катаракты, удаляют смещаемый хрусталик, заменяя его на искусственный.
- При функциональной дисфункции опорно-двигательного аппарата возникает необходимость стабилизирования позвоночника с помощью металлических пластин.
- При выраженной деформировании грудной клетки проводится торакопластика.
- При протрузии тазобедренных суставов производят внутреннее протезирование суставов.
Прогноз
Длительность жизни в среднем при синдроме Марфана составляет 30-45 лет.
Известно, что это многие знаменитые личности страдали данным синдромом. Это и Ганс Христиан Андерсен – датский писатель, автор знаменитой Русалочки; Авраам Линкольн – 16 президент США, Майкл Феллпс- известный пловец, многократный олимпийский чемпион. А также известные композиторы – Никколо Паганини, Сергей Рахманинов.
Люди с данной патологией должны тщательно следить за своим здоровьем, постоянно наблюдаться и консультироваться со своим лечащим врачом, избегать чрезмерных физических нагрузок.
По мимо медикаментозного лечения, необходимо проведение профилактических мероприятий с целью улучшения общего самочувствия, повышения иммунитета, соответствующий режим труда и отдыха.
Видеозаписи по теме
Интересное

Высшее образование (Кардиология). Врач-кардиолог, терапевт, врач функциональной диагностики. Хорошо разбираюсь в диагностике и терапии заболеваний дыхательной системы, желудочно-кишечного тракта и сердечно-сосудистой системы. Закончила академию (очно), за плечами большой опыт работ.Специальность: Кардиолог, Терапевт, Врач функциональной диагностики. .
Поделиться: